很漂亮的两篇文章,不论是站在筛选部、还是药化、计算部,亦或是生物部,都可圈可点,直接指向了 FIC 药物开发的几大核心问题[1-2]:
- 药物应该靶向蛋白质的哪个结构域? 计算这个时候能发挥作用么?
- 采用什么方式发现苗头化合物?方法和策略不同,注定了 Novartis 和 Vividion 化合物效果的差异性。
- 发现了系列苗头化合物,选择哪个化学系列重点推进?怎么快速提高化合物的整体性能,尤其是像降低logP和提高渗透性这种似乎矛盾的地方,让其快速成为药!
一、WRN Helicase domian is the KEY!
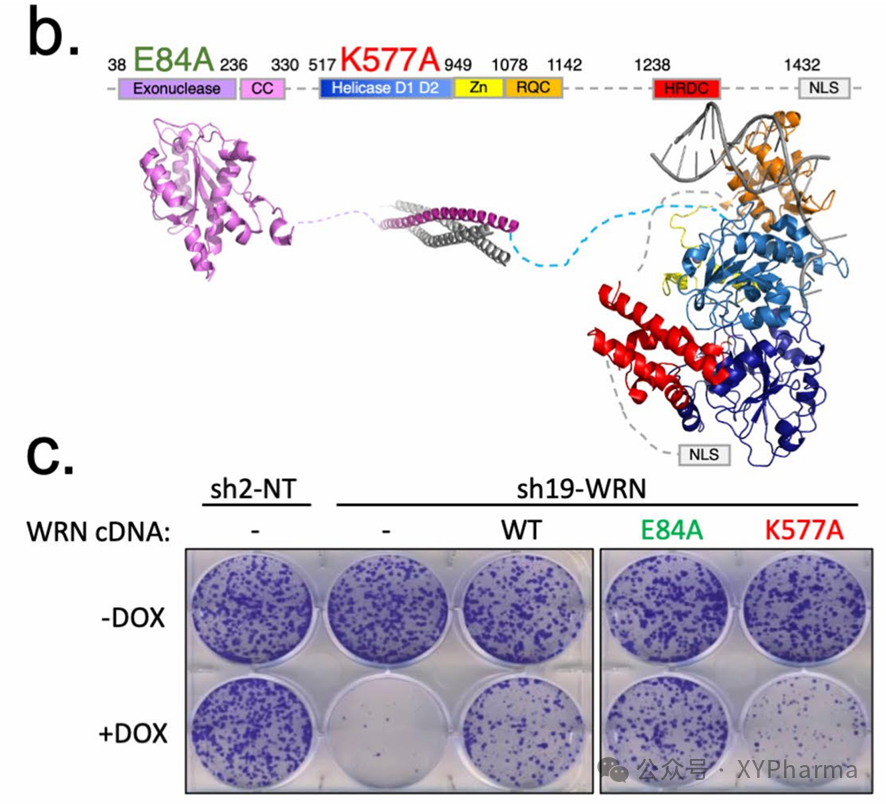
WRN 有几个不同的结构域,如果是抑制其功能,抑制哪一部分才能在 MSI 中发挥作用?如果确定了核心发挥药理作用的结构域,采用什么方法发现 Hits?

对于第一个问题,将 WRN 突变,使其丧失部分功能,或者 knockdown/knockout 等,检测下活性,再复表达能完成验证。
通过 kd 和复表达,Novartis 确定了在 MSI 和 WRN 表现的合成致死仅和 Helicase 解螺旋功能有关。
二、采用什么药物筛选方式?
接下来就是第二个问题,怎么发现苗头化合物?
Follow 的项目有前人踏路还好,但是 FIC 项目,只能自己摸索。如果有计算部门,直接丢给计算人员,让计算人员去建模、虚拟筛选、计算结合能、分子动力学模拟·······然后测试活性,那计算部门算是头疼了。
虽然确定了核心是解螺旋结构域,但是是选择 ATP 的结合部位,还是核酸结合部位,抑或是别构部位?核酸结合部位太浅,ATP 结合部位有 walker motif,结合的驱动力是由于静电作用。在分析氨基酸的保守性,发现可能难以实现选择性。
那就定别构,但是定哪里的别构?把一个静态蛋白的各个口袋全部框选出来?每个口袋进行对接筛选?不是头呀!如果再碰到领导固执,坚持在现阶段就虚拟筛选,验证下花几十万招的计算人员能力,验证花大几十万买的药物设计软件,那就只能逼走计算人员出去开公司拉融资当老板……
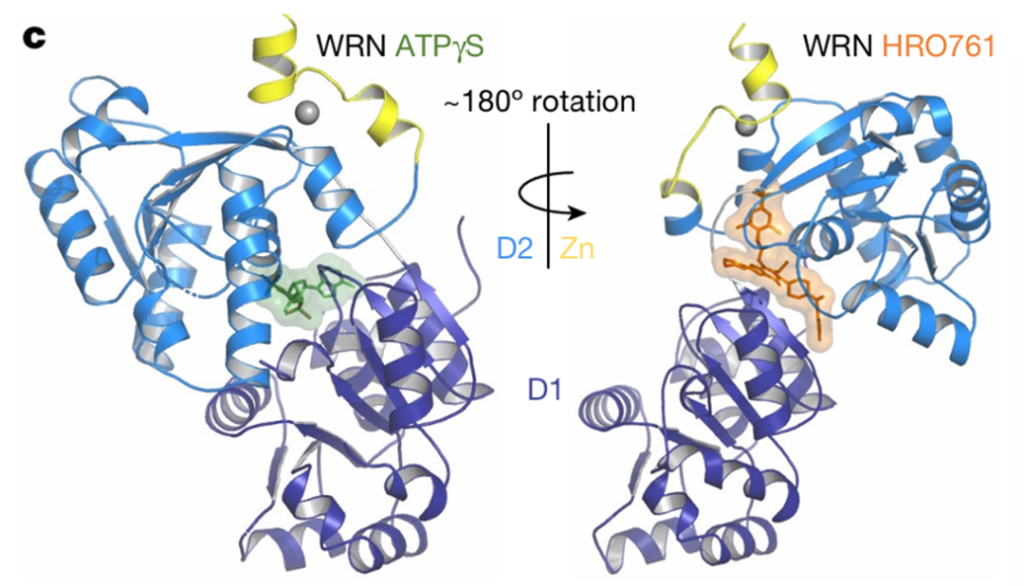
看诺华的最终晶体解析数据,左侧的为 ATP 类似物和解螺旋结构域的结合构象,右侧是和抑制剂的结构构象,其中部分结构直接就反转了 180°,这个是计算不出来的……
终于开始进行表型筛选,检测 ATP 依赖性的 DNA 解螺旋和 DNA 依赖性的 ATP 酶活性,有试剂盒似乎并没有什么问题。然后,生物部开始进行筛选,结果发现命中率特别低。这个时候,领导怀疑生物人员的水平,详细询问操作的各个步骤细节,没发现问题,又说先检测下已有的 WRN 抑制剂,验证下试验人员检测准确性。结果发现,这个人检测的出来,那个人检测不出来的,重复后,两个人又反过来了。生物部开始抓狂,抠死各种细节,终于确定了能够维持稳定性的方法。
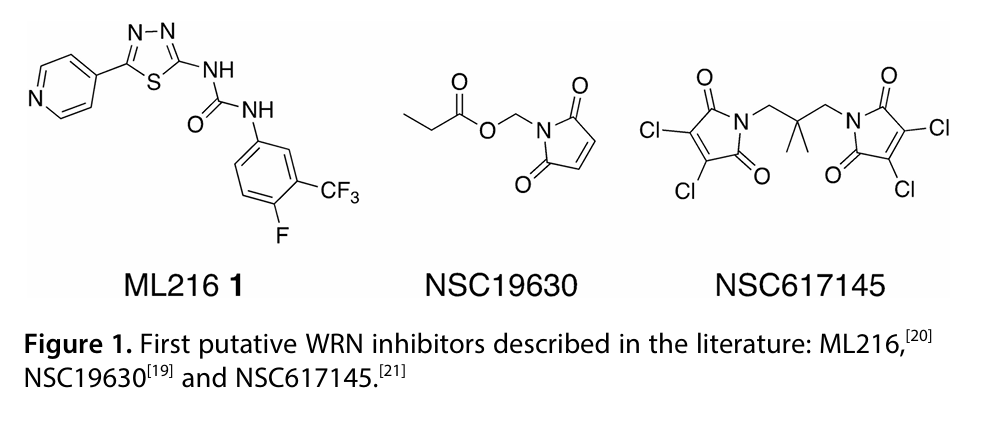
后面发现检测方法是稳定了,但是似乎公认的 WRN 抑制剂或许根本从头就是错的,比如化合物可能会诱导的蛋白聚集或者沉淀,亦或者化合物会和核酸作用等各种作用。。。
诺华直接出了一篇 ChemMedChem 来记录吐槽这段儿艰辛的探索之路 [3]。
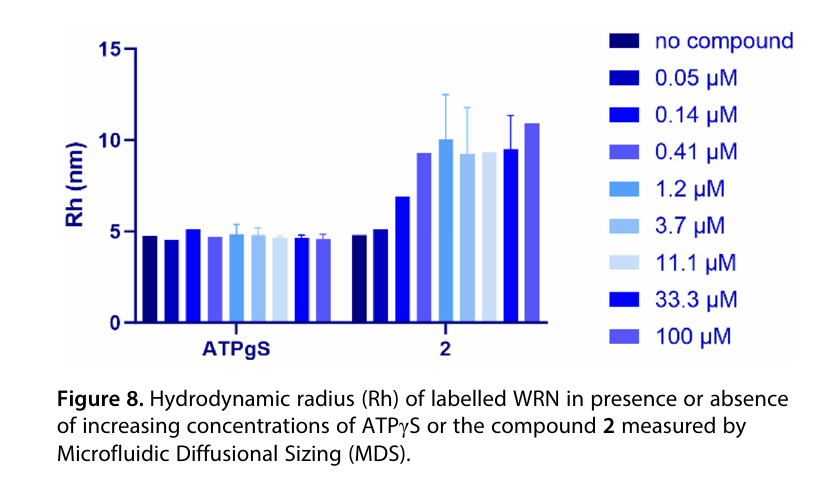
后面整合团队,深入分析,终于确定了合适的筛选方法。左侧的是 Novartis 采用的筛选验证方式,右侧是 Vividion 采用的方式。
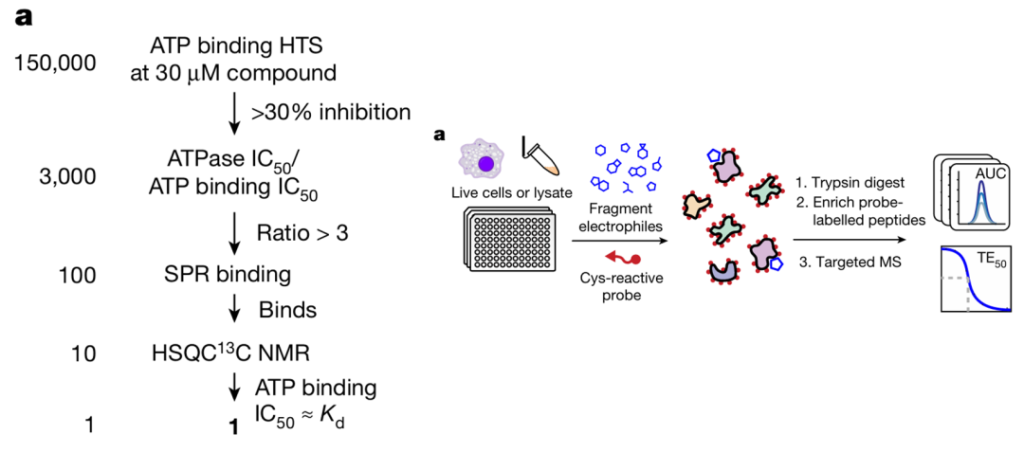
Novartis 部分:最开始采用通用的方式,WRN helicase assay,进行 ATP 酶活性分析,发现假阳性太高,命中也只发现共价化合物。进一步确定,发现是一个 ATP 竞争性的靶向 Cys727 的别构共价抑制剂。后面为了发现非共价抑制剂,采用的是 ATP-binding assay,(Cy5-ATPgamaS binding assay using TR-FRET)。
TR-FRET(时间分辨荧光共振能量转移)是一种基于FRET(荧光共振能量转移)的生物化学检测技术,通常用于研究分子间的相互作用和距离变化。这种技术的核心在于两个荧光标记分子之间的能量转移,其机制如下:1. 荧光标记实验中需要两种荧光分子,一种是供体(donor),另一种是受体(acceptor)。在本实验中,铕标记的链霉亲和素作为供体,而Cy5标记的ATPγS作为受体。
Vividion 部分:左侧为ADP,右侧为共价结合的抑制剂,和原始 ATP/ADP 结合构象大概类似。
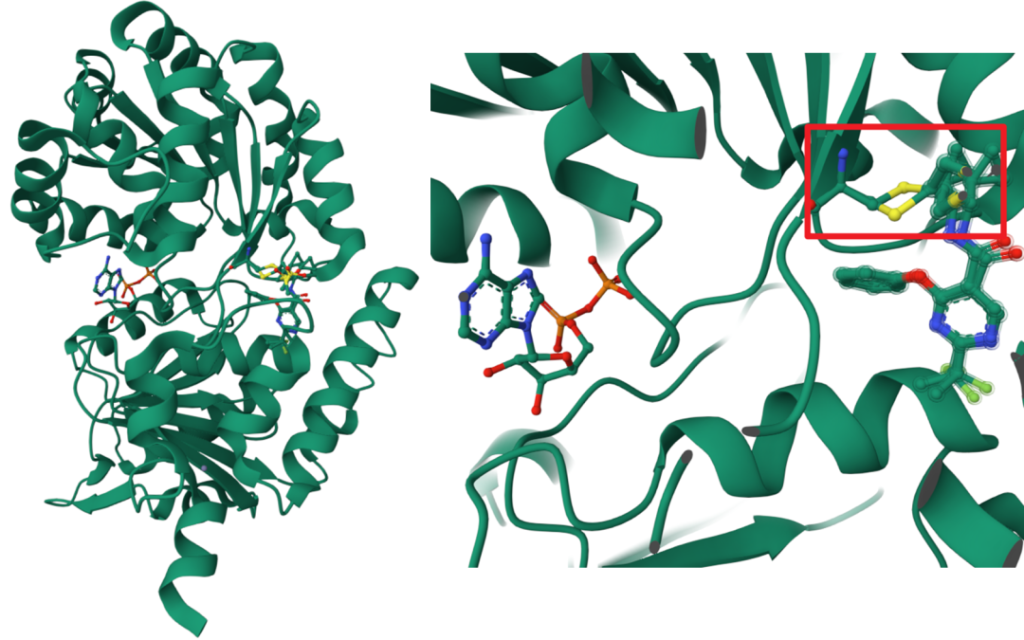
三、结构优化
千辛万苦走到了这一步,药化和合成开始发力,设计合成各种化合物,提高活性,提高类药性,克服种种困难。可竞争对手开始报道了大额融资,竞争对手的专利公开,竞争对手拿到了 IND。我的天!!!
领导开始说他们怎么这么快?!合成说和我没关系,兄弟们为了项目进度,天天加班,都睡在公司了。药化说这个结构有问题,提高了溶解度,口服吸收没了。口服吸收好了,活性没了。领导说,那为什么人家跑到前面去了,我不管,我只要结果。
好在有了晶体结构解析,计算部的作用就可以发挥了,基于结合口袋进行片段生长和替换,生成了大量的分子。然后在基于分子的物理性质预测,希望能够在矛盾的性质中发现“太极”结构 [4]。
Novartis 部分:
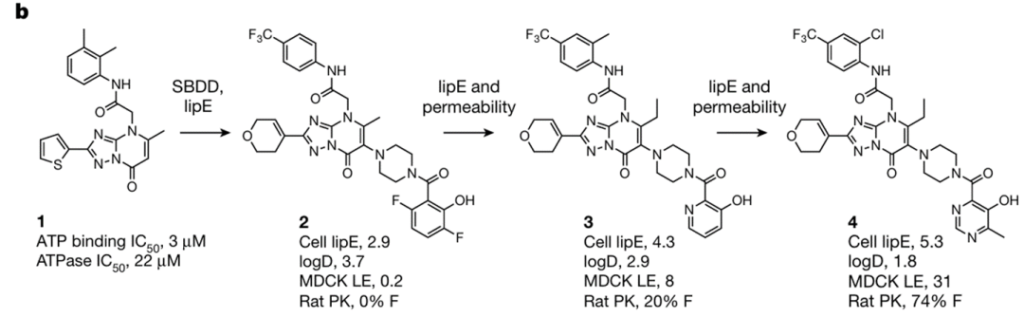
终于:Two examples of transformations that lower logP while improving permeability are the introduction of the ortho-methyl aniline in compound 3 and the hydroxy pyrimidine in compound 4, HRO761.
最后,计算证明了自己,药化调整可合成性,和合成兄弟搭伙继续加班睡公司。
Vividion 部分:
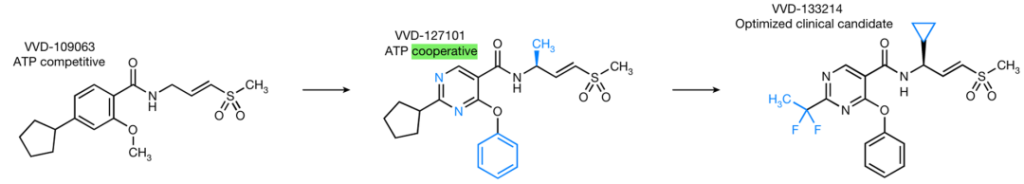
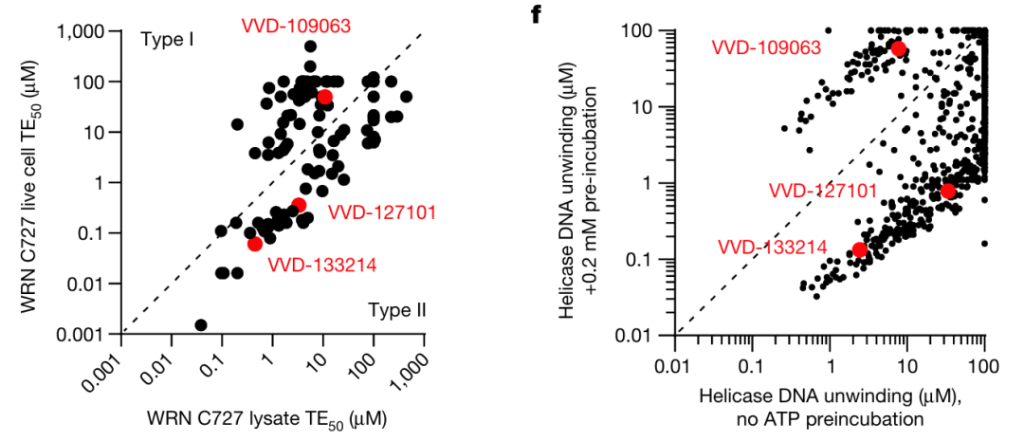
分析生物数据的时候,发现一点儿异常,左图是 Target engagement,右侧是 DNA unwinding 数据。左图中,项目负责人发现了异常,似乎在活细胞和裂解液中测量靶点的占据率会有两类分子,进一步分析可能是由于裂解液中存在多次稀释,所以 ATP 浓度低了。
右图继续验证,横坐标是没有 ATP,纵坐标为有 ATP 预孵育,立马发现了区别,对角线以上 VVD-109060 系列明显是 ATP 竞争性的,但是下方 VVD-127101 却是协同性的,在ATP 存在下,更好了。最后,Vvidion 采用了 101 系列进一步推进。